Make2Discover
This study introduces a low-cost, open-source, semiautomated bioreactor system, developed using Arduino and Raspberry Pi, for longitudinal microbial culture studies. The system protocol includes detailed instructions for custom-designed components, specifications for easily purchasable parts, and open-source code for a web-based control interface. It was validated for sterility and tested in a case study involving the cultivation of Phanerochaete chrysosporium and Trichoderma reesei to assess their metabolic profile, both in isolation and coculture, for lignin degradation. Using mass spectrometry coupled with gas chromatography, several lignin degradation intermediates were annotated, including 4-hydroxybenzoic acid, vanillic acid, and ferulic acid, along with their temporal detection over 25 days. Additionally, we developed an annotation workflow to search for enzymatic functions producing these compounds in the genomes of P. chrysosporium and T. reesei, providing multiple layers of evidence to describe, for the first time, a computational annotation for lignin metabolism in these fungi. The results highlighted the production of lignin breakdown intermediates by the two fungal species, with annotations of the respective enzymatic functions, while also demonstrating the bioreactor’s flexibility and suitability for diverse biotechnological applications, particularly in the field of biodegradation and waste valorization.

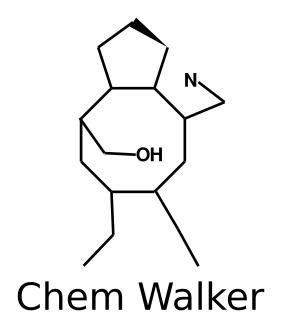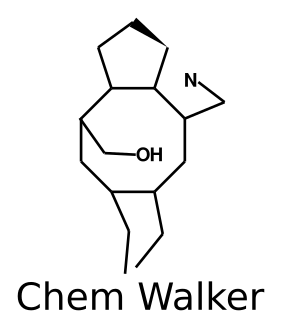
Annotation propagation through random walks
Annotation of the mass signals is still the biggest bottleneck for the untargeted mass spectrometry analysis of complex mixtures. Molecular networks are being increasingly adopted by the mass spectrometry community as a tool to annotate large-scale experiments. We have previously shown that the process of propagating annotations from spectral library matches on molecular networks can be automated using Network Annotation Propagation (NAP). One of the limitations of NAP is that the information for the spectral matches is only propagated locally, to the first neighbor of a spectral match. Here, we show that annotation propagation can be expanded to nodes not directly connected to spectral matches using random walks on graphs, introducing the ChemWalker python library.
Explainable artificial intelligence
Antimicrobial resistance (AMR) is one of the most concerning modern threats as it places a greater burden on health systems than HIV and malaria combined. Current surveillance strategies for tracking antimicrobial resistance (AMR) rely on genomic comparisons and depend on sequence alignment with strict similarity cutoffs of greater than 95%. Therefore, these methods have high false-negative error rates due to a lack of reference sequences with a representative coverage of AMR protein diversity. Deep learning has been used as an alternative to sequence alignment, as artificial neural networks can extract abstract features from data, thereby limiting the need for sequence comparisons. Here, a convolutional neural network (CNN) was trained to differentiate between antimicrobial resistance proteins and non-resistance proteins, and to annotate them in nine resistance classes. Our model demonstrated higher recall values (> 0.9) than the alignment-based approach for all protein classes tested. Additionally, our CNN architecture allowed us to investigate internal states and explain the model classification regarding protein domain feature importance related to antimicrobial molecule inactivation. Finally, we built an open-source bioinformatic tool (https://github.com/computational-chemical-biology/DeepSEA-project) that can be used to annotate antimicrobial resistance proteins and provide information on protein domains without sequence alignment.
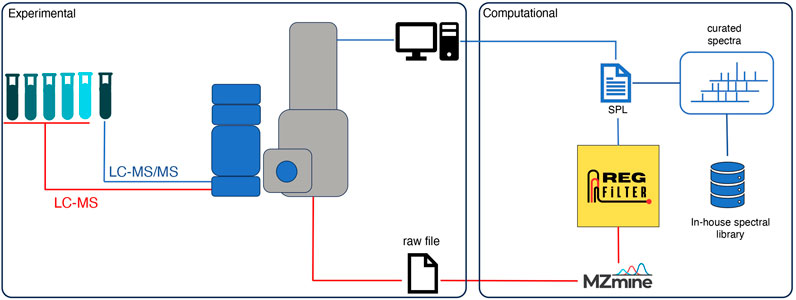
Semi-automated feature selection
Untargeted metabolomics is often used in studies that aim to trace the metabolic profile in a broad context, with the data-dependent acquisition (DDA) mode being the most commonly used method. However, this approach has the limitation that not all detected ions are fragmented in the data acquisition process, in addition to the lack of specificity regarding the process of fragmentation of biological signals. The present work aims to extend the detection of biological signals and contribute to overcoming the fragmentation limits of the DDA mode with a dynamic procedure that combines experimental and in silico approaches.